


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
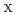 (X = 1, 2)
(X = 1, 2)城戸 健太朗
International Journal of Quantum Chemistry, 121(21), p.e26781_1 - e26781_15, 2021/11
被引用回数:1 パーセンタイル:17.5(Chemistry, Physical)Ruthenium tetroxide (RuO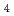 ) is one of chemical species of fission products assumed to be released to the environment during a severe accident of nuclear facilities and a target compound to assess the amount produced, reactivity, mobility and release timing. In this article, the NO (X = 1, 2) adduct formation of RuO has been investigated, based on the potential energy curve (PEC) evaluated by UM06, UTPSSh, CASSCF, and CASPT2 methods. At several stationary points, CCSD and LR-CCSD(T) energies are also computed for a comparison. The PEC shows that there is an activation barrier to form the NO adduct and that the process is endothermic in terms of free energy. In the system, the electron transfer occurs from NO to RuO when the bond between the nitrogen and oxo ligand is formed. It has been discussed in detail using active orbitals, weight of electron configurations and spin population obtained by CASSCF.
) is one of chemical species of fission products assumed to be released to the environment during a severe accident of nuclear facilities and a target compound to assess the amount produced, reactivity, mobility and release timing. In this article, the NO (X = 1, 2) adduct formation of RuO has been investigated, based on the potential energy curve (PEC) evaluated by UM06, UTPSSh, CASSCF, and CASPT2 methods. At several stationary points, CCSD and LR-CCSD(T) energies are also computed for a comparison. The PEC shows that there is an activation barrier to form the NO adduct and that the process is endothermic in terms of free energy. In the system, the electron transfer occurs from NO to RuO when the bond between the nitrogen and oxo ligand is formed. It has been discussed in detail using active orbitals, weight of electron configurations and spin population obtained by CASSCF.
宮原 直哉; 三輪 周平; 堀口 直樹; 佐藤 勇*; 逢坂 正彦
Journal of Nuclear Science and Technology, 56(2), p.228 - 240, 2019/02
被引用回数:7 パーセンタイル:61.94(Nuclear Science & Technology)軽水炉シビアアクシデント時のソースターム評価における核分裂生成物(FP)化学挙動評価モデルを高度化するため、FP化学データベース「ECUME」の初版を構築した。ECUMEには、代表的な事故シーケンスにおける主要な化学反応と、その実効的な化学反応速度定数を実装する計画である。初版においては、300-3000Kの温度領域におけるCs-I-B-Mo-O-H系の主要化学種に対し、それらの生成に係る化学反応の速度定数を文献調査または第一原理に基づく理論計算によって整備した。構築した化学反応データセットを用いた解析の一例として化学反応解析を実施した結果、1000Kにおいて有意な化学反応速度の効果が見られた。また、平衡に至った後の化学組成を化学平衡計算の結果と比較したところ、代表的なCs-I-B-Mo-O-H系化学種に対して良く整合する結果が得られた。これらの結果から、構築したデータセットは、速度論の考慮が必要なシビアアクシデント時のCs-I-B-Mo-O-H系FP化学挙動評価のために有用であるとの結論を得た。
 system
system黒崎 譲; 高柳 敏幸*
Chemical Physics Letters, 406(1-3), p.121 - 125, 2005/04
BrH系の非経験的に計算された基底状態のグローバルなポテンシャルエネルギー面(PES)についての新しい解析的関数を構築した。これは、以前われわれが発表した1 A' PES[Y. Kurosaki, T. Takayanagi, J. Chem. Phys. 119 (2003) 7383]の修正版である。反応H+HBr
A' PES[Y. Kurosaki, T. Takayanagi, J. Chem. Phys. 119 (2003) 7383]の修正版である。反応H+HBr H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol
H+Brとその同位体置換した反応の速度定数を、新しい1A' PESを用いて計算したところ実測値をよく再現した。これはフィットした関数の反応障壁の値1.53kcal mol が真の値に非常に近いことを強く示唆している。
が真の値に非常に近いことを強く示唆している。
黒崎 譲; 横山 啓一; 寺西 慶哲
Chemical Physics, 308(3), p.325 - 334, 2005/01
被引用回数:23 パーセンタイル:60.47(Chemistry, Physical)248及び193nmの光によるギ酸の二つの解離反応、HCOOHHO+CO(1)及びHCOOHCO+H(2)、について、RMP2(full)/cc-pVDZレベルでのdirect ab initio molecular dynamics法を用いて約1200本の古典トラジェクトリを計算した。その結果、反応2における相対並進モードへのエネルギー分配の割合は、反応1におけるそれよりもはるかに大きいことが明らかとなった。このことは反応1と2の遷移状態の構造の違いによるところが大きいと考えられる。また、生成物であるCOとHはともに振動,回転状態が励起されていることが予測された。本計算から見積もられたCOの振動状態分布は、248nm光による光解離実験の値とよく一致した。
 potential energy surfaces for the lowest three doublet states (1A', 2A', and 1A") of the BrH system
potential energy surfaces for the lowest three doublet states (1A', 2A', and 1A") of the BrH system黒崎 譲; 高柳 敏幸
Journal of Chemical Physics, 119(15), p.7838 - 7856, 2003/10
被引用回数:26 パーセンタイル:63.68(Chemistry, Physical)BrH系の三つの最低二重項状態(1A', 2A', 1A") についての断熱ポテンシャルエネルギー面を、Breit-Pauliハミルトニアンに基づくスピン-軌道相互作用による補正を加えてMRCI/aug-cc-pVTZ法により計算し、得られた断熱エネルギーを解析的な多体関数にフィットした。基底状態のポテンシャル面上での水素引抜き及び水素交換の反応障壁は、MRCI+Q/aug-cc-pVTZレベルでそれぞれ1.28 and 11.71 kcal molと計算された。ポテンシャル面のフィットの精度は0.1 kcal mol以内であった。フィットした基底状態のポテンシャル面を用いて、水素引抜き及び水素交換並びに同位体置換した反応の反応速度定数を、変分的遷移状態理論に基づくICVT/LAG法により計算した。その結果、水素引抜きについては実験との一致はおおむね良好であったが、水素交換については計算値は実験値を大幅に下回った。この不一致は、実験データの不足が主な原因と考えられる。
宮崎 幹也*
JAERI-Data/Code 2003-007, 55 Pages, 2003/05
 JAERI-Data-Code-2003-007.pdf:2.53MB
JAERI-Data-Code-2003-007.pdf:2.53MB
新物質や新デバイスなどの研究開発は、これからの社会の発展を支える中心的な役割を果たすと考えられているが、経験的・実験的なアプローチが主体となったこれまでの取り組みは、もはや限界に達している。今後は物質の構造をより微細な状態で取り扱う必要があり、これまでの古典的な理論を用いた計算手法では既に、精度上の限界が生じている。これらの問題を解決する手法として、量子力学に基礎を置く最先端の電子状態計算手法についての研究が進められている。第一原理計算による物性予測に基づいた、このマテリアルデザインの手法は、このような状況におけるブレークスルーとなる可能性が極めて高いと考えられている。本報告書は、2002年9月17~21日にかけて原研ITLB棟及び国際高等研で行われた、「コンピュテーショナル・マテリアルズ・デザイン ワークショップ」(大阪大学「計算機ナノマテリアルデザイン」プロジェクト主催)において講習を実施したMACHIKANEYAMA-2000及びOSAKA-2000をPCクラスタシステムに導入した際に行った、サンプル計算問題を用いた動作確認の結果についてまとめたものである。さらに、これらの計算コードの利用環境整備の一環として、Graphical User Interface(GUI)環境の整備について検討を行った。
 CHOCH+HCO; Direct ab initio molecular dynamics study
CHOCH+HCO; Direct ab initio molecular dynamics study黒崎 譲; 横山 啓一
Chemical Physics Letters, 371(5-6), p.568 - 575, 2003/04
被引用回数:31 パーセンタイル:69.48(Chemistry, Physical)UB3LYP/cc-pVDZレベルでの直接非経験的分子動力学法を用いて、T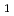 ポテンシャル面上での光分解反応,CHCHOCH+HCO、について全部で400本のトラジェクトリを計算した。その結果、反応生成物であるCHは振動,回転ともに励起しないが、HCOは振動励起しないものの回転励起することが予測された。トラジェクトリ計算の結果を平均するとHCOの回転エネルギーは1.1kcal/molとなり、これは利用可能なエネルギー、7.3kcal/molの15.1%にあたる。本計算結果は実測値と数%の誤差で一致している。
ポテンシャル面上での光分解反応,CHCHOCH+HCO、について全部で400本のトラジェクトリを計算した。その結果、反応生成物であるCHは振動,回転ともに励起しないが、HCOは振動励起しないものの回転励起することが予測された。トラジェクトリ計算の結果を平均するとHCOの回転エネルギーは1.1kcal/molとなり、これは利用可能なエネルギー、7.3kcal/molの15.1%にあたる。本計算結果は実測値と数%の誤差で一致している。
 study of hydrogen hydrate clathrates for hydrogen storage within the ITBL environment
study of hydrogen hydrate clathrates for hydrogen storage within the ITBL environmentSluiter, M. H. F.*; Belosludov, R. V.*; Jain, A.*; Belosludov, V. R.*; 安達 斉*; 川添 良幸*; 樋口 健二; 大谷 孝之
Lecture Notes in Computer Science 2858, p.330 - 341, 2003/00
「水素分子が氷に大量に吸蔵されメタンと同様のクラスレートを形成する可能性がある」という実験的報告がある。構造はメタン吸蔵クラスレートと同様であり、水素ガスの500倍もの濃度で吸蔵がなされるという。ところが、実験的にはX線や中性子線を使っても水素分子の吸蔵位置や結合状況の詳細を決定できていなかった。われわれは、この結果に興味を持ち、その構造と物性の詳細を第一原理シミュレーション計算によって研究することにした。しかしながら、その計算には大規模な計算能力が不可欠である。このため、原研が開発しているITBL基盤技術を活用することによって複数台のスーパーコンピュータを連携し、大規模な第一原理計算を実現した。
CHOCH +CO: Direct ab initio dynamics study
+CO: Direct ab initio dynamics study黒崎 譲; 横山 啓一
Journal of Physical Chemistry A, 106(47), p.11415 - 11421, 2002/11
被引用回数:36 パーセンタイル:73.04(Chemistry, Physical)RMP2(full)/cc-pVDZレベルでのdirect ab initioダイナミクス法を用い、S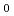 面上での光解離反応CHCHOCH+COのトラジェクトリを全部で100個計算した。相対並進エネルギー,CO内部エネルギー,CH内部エネルギーに対するエネルギー分配はそれぞれ、28,20,51%と計算された。生成物のCOは高回転励起状態に励起されるが、振動状態は励起されないことが予測され、平均の回転及び振動量子数はそれぞれ68.2,0.15と計算された。この結果は、Ghermanらの実験(J.Chem.Phys.2001, 114, 6128)と定性的に一致している。
面上での光解離反応CHCHOCH+COのトラジェクトリを全部で100個計算した。相対並進エネルギー,CO内部エネルギー,CH内部エネルギーに対するエネルギー分配はそれぞれ、28,20,51%と計算された。生成物のCOは高回転励起状態に励起されるが、振動状態は励起されないことが予測され、平均の回転及び振動量子数はそれぞれ68.2,0.15と計算された。この結果は、Ghermanらの実験(J.Chem.Phys.2001, 114, 6128)と定性的に一致している。
H + Cl CHCl + Cl reaction; Ab initio molecular orbital study黒崎 譲
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
反応CH + Cl CHCl + Clのポテンシャルエネルギー面を、CASSCF及びMRCIレベルでcc-pVTZ基底関数を用いて計算した。その結果、この反応はCHCl + Cl サイドから見て、CASSCFレベルでは小さな反応障壁をもつが、MRCIレベルでは反応障壁を持たないことが明らかとなった。すなわち、反応CHCl + Cl CH + Clは自発的な反応であることが予測された。MRCI計算の結果は、以前われわれが行ったPMP4(SDTQ)レベルでの計算結果 [J. Mol. Struct. (Theochem) 503 (2000) 231]を強く支持するものである。
at 157.6nm, 2; A Theoretical study using wave packet propagation横山 啓一; 横山 淳; 高柳 敏幸
Journal of Chemical Physics, 114(4), p.1624 - 1630, 2001/01
被引用回数:4 パーセンタイル:12.25(Chemistry, Physical)新しいレーザー同位体分離手法の開発につながる結合選択的光分解反応の例として、CBrClFの波長157.6nmにおける真空紫外光分解の動力学を波束の時間発展を量子学的に計算することにより調べた。実験的に観測されている3体解離反応(CBrClF→Br+Cl+CF)の存在を理論的に確認し、波束の運動を通してその動力学的機構を明らかにした。
中沢 哲也; 横山 啓一; Grismanous, J.*; 片野 吉男*
Journal of Nuclear Materials, 279(2-3), p.201 - 206, 2000/06
被引用回数:10 パーセンタイル:57.14(Materials Science, Multidisciplinary)本論文ではシリカ表面水酸基の化学的特性に対する他元素(B,Al,Ga)の添加効果や非架橋酸素に配位したリチウム原子の影響を調べた。種々のリチウムシリケイト表面クラスターモデルを用いて表面水素原子がH として脱離するのに必要なエネルギーと電子状態に対して分子軌道計算を行い検討した。その結果、表面水素原子のイオン性は添加原子の表面酸素への直接相互作用によって強まり、Li原子の非架橋酸素への配位によって弱まることがわかった。また、表面水素原子の脱離エネルギーはそのイオン性の増加に伴い減少することがわかった。
として脱離するのに必要なエネルギーと電子状態に対して分子軌道計算を行い検討した。その結果、表面水素原子のイオン性は添加原子の表面酸素への直接相互作用によって強まり、Li原子の非架橋酸素への配位によって弱まることがわかった。また、表面水素原子の脱離エネルギーはそのイオン性の増加に伴い減少することがわかった。
鈴木 篤之*; 長崎 晋也*
JNC TJ1400 99-028, 62 Pages, 1999/02
前半部では、非結晶性鉄酸化物コロイド粒子へのNpO/SUB2/SUP+の吸着拳動に関して吸着平衡と吸着速度という2つの観点から検討を加えた。その結果、吸着挙動はバルク溶液とコロイド粒子外表面間の遠い吸着と、コロイド粒子内のマイクロポアに拡散し吸着する遅い吸着の2つのステップから構成されることを明らかにした。また、外表面へのNpO/SUB2/SUP+の吸着が内圏型吸着であること、マイクロポア内の表面拡散係数が2.0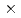 10/SUP-13cm/SUP2/Sであることを示した。後半部では、ab initio計算法を用い、数種類のウラニル錯体の振動数を評価しラマン分光などの実測結果と比較するとともにウラニルイオンの水和の影響について考察を行った。また、銀コロイド粒子へのウラニルイオンの吸着挙動の解析を行い、ラマン分光法の実験結果とも合わせて、内圏型での吸着の可能性が高くそのときの銀-ウラン原子間距離が3A○であると評価した。
10/SUP-13cm/SUP2/Sであることを示した。後半部では、ab initio計算法を用い、数種類のウラニル錯体の振動数を評価しラマン分光などの実測結果と比較するとともにウラニルイオンの水和の影響について考察を行った。また、銀コロイド粒子へのウラニルイオンの吸着挙動の解析を行い、ラマン分光法の実験結果とも合わせて、内圏型での吸着の可能性が高くそのときの銀-ウラン原子間距離が3A○であると評価した。
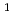 A' electronic states of H
A' electronic states of H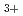
市原 晃; 横山 啓一; 岩本 修
JAERI-Data/Code 98-031, 18 Pages, 1998/11
JAERI-Data-Code-98-031.pdf:0.89MB
基底及び第一電子励起A'状態におけるHのポテンシャルエネルギーを701の異なる空間構造に対して計算し、その結果を表にまとめた。これらのポテンシャルエネルギーは、[8s6p2d1f]ガウス型基底関数を用いた、非経験的分子軌道論に基づく完全な配置間相互作用法により計算された。ポテンシャルの計算値を内挿することにより、二つのポテンシャル面間のavoided crossingや基底状態ポテンシャル面のエネルギー極小値付近の形状を表現することが可能である。ポテンシャルエネルギーは三つの核間距離の関数として0.6から10.0ボーアの範囲内で与えられており、H+H衝突に関する分子動力学の研究に適している。
池添 康正; 鈴木 和弥; 中島 幹雄; 横山 淳; 白石 浩二; 大野 新一*
JAERI-Research 98-051, 43 Pages, 1998/09
JAERI-Research-98-051.pdf:1.63MB
アンモニアクラスターイオン(NH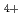 .nNH)について、非経験的分子軌道法計算による解析及びコロナ放電-Jet expansion法による生成・分解実験を行った。分子軌道計算はGaussian94を用いて、最適構造、全電子エネルギー、基準振動の振動数を求めた。クラスター生成については、放電電流、ガス組成と圧力、ガス噴出細孔の径等のクラスターサイズ分布に対する効果を調べた。ガス噴出細孔部におけるクラスター成長はクラスターサイズn単位で1以下であった。クラスターの熱分解については、放電電流、細孔径、飛行時間、クラスターサイズによる分解速度の変化を調べた。実験結果をもとにして、クラスターによる分解速度の変化を調べた。実験結果から、クラスターの内部エネルギーの多寡に主たる寄与をする過程は、クラスター生成、分解反応であることを推論した。
.nNH)について、非経験的分子軌道法計算による解析及びコロナ放電-Jet expansion法による生成・分解実験を行った。分子軌道計算はGaussian94を用いて、最適構造、全電子エネルギー、基準振動の振動数を求めた。クラスター生成については、放電電流、ガス組成と圧力、ガス噴出細孔の径等のクラスターサイズ分布に対する効果を調べた。ガス噴出細孔部におけるクラスター成長はクラスターサイズn単位で1以下であった。クラスターの熱分解については、放電電流、細孔径、飛行時間、クラスターサイズによる分解速度の変化を調べた。実験結果をもとにして、クラスターによる分解速度の変化を調べた。実験結果から、クラスターの内部エネルギーの多寡に主たる寄与をする過程は、クラスター生成、分解反応であることを推論した。
中沢 哲也; 横山 啓一; 野田 健治
Journal of Nuclear Materials, 258-263, p.571 - 575, 1998/00
被引用回数:14 パーセンタイル:73.06(Materials Science, Multidisciplinary)リチウムシリケイト及びAl添加リチウムシリケイトは核融合炉用固体増殖材の候補材料である。本研究では、これら材料表面に存在する水素原子の化学的性質を非経験的分子軌道計算により調べた。その結果、表面水素のイオン性がAl原子の表面酸素への相互作用によって強まる。その一方で、非架橋酸素に配位するLiイオンによってそのイオン性は弱まることが示された。また、表面酸素に対する表面水素の親和力は表面水素のイオン性の増加とともに低下することが示された。
loss reaction from ethane cation, CH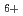
黒崎 譲*; 高柳 敏幸
Chemical Physics Letters, 277(4), p.291 - 298, 1997/00
被引用回数:10 パーセンタイル:35.52(Chemistry, Physical)エタンカチオンの水素分子脱離反応に対して非経験的分子軌道計算を行った。その結果、2つの遷移状態(TS)と2つの中間生成物(IP)が発見された。極限的反応座標(IRC)計算により、これらのTSとIPは1つの反応経路上にあることが確認された。また、RRKM法によって反応速度を見積もったところ、トンネル効果はあまり大きくないことが予測された。この反応速度の計算結果は、これまでの実験結果とよく一致していることが確認された。
system with the trajectory-surface-hopping model市原 晃; 白井 稔三; 横山 啓一
Journal of Chemical Physics, 105(5), p.1857 - 1861, 1996/08
被引用回数:38 パーセンタイル:78.01(Chemistry, Physical)非経験的分子軌道計算(ab initio full configuration interaction calculation with a [8s6p2d1f] Gaussian type basis set)で得られたHの3次元ポテンシャル面上でtrajectory-surface-hopping法を用いることにより、D+H、D+D、及びH+D衝突で生じる電荷移動及び粒子組み替え反応の断面積を計算した。ポテンシャル面間の非断熱的電子遷移の確率は、Landau-Zener-Stuckelberg近似に基づいて評価した。その結果、衝突エネルギーが2.5eVから5.0eVの範囲では、計算結果は実験値と定量的に一致することが確認された。
S+O( P)HS+OH reaction; A Theoretical study
P)HS+OH reaction; A Theoretical study横山 啓一; 高柳 敏幸
Journal of Chemical Physics, 104(5), p.1953 - 1957, 1996/02
被引用回数:5 パーセンタイル:21.08(Chemistry, Physical)トンネリングは化学反応の動力学を支配する重要な因子の1つである。トンネリングは正当に評価できる量子論的手法によりトンネル効果を取り入れた反応速度定数を計算することができる。今回はHS+O(P)HS+OHの反応を例にとり、時間に依存しないシュレディンガー方程式を数値的に解くことによって反応速度を計算し実測値と比較した。計算に用いたポテンシャルエネルギー面をわずかに修正することにより実測の温度依存性を再現できた。さらに、トンネリングを考慮しない単純な遷移状態理論から導かれる速度定数を比較することにより、トンネリングが室温付近でも100倍程度、反応を加速することが明らかになった。
CN molecule; An Experimental and ab initio study橋本 雅史; 横山 啓一; 工藤 博司*; Wu, C. H.*; P.von-R.Schleyer*
Journal of Physical Chemistry, 100(39), p.15770 - 15773, 1996/00
被引用回数:13 パーセンタイル:43.8(Chemistry, Physical)高温質量分析法により、金属ナトリウムNaCNとの混合試料上の平衡蒸気中に超原子価分子NaCNの存在を確認し、その熱力学量を決定した。また同時にab initio MO計算により分子の安定構造およびエネルギー状態についての理論値を得た。NaCNの解離エネルギー(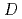
 (NaCN-Na)は、実験値は24.8
(NaCN-Na)は、実験値は24.8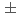 1.6kcal/mol、理論値は17.4kcal/molとなり、この分子が熱力学的に安定であることを示している。NaCN分子のイオン化エネルギーは実験値4.920.2ならびに理論値4.66eVを得た。理論計算により、NaCN分子は平面2個直線2個の計4個の安定構造を持ち、最も安定な構造は平面型であることがわかった。前回報告したLiCN分子との比較を混じえ、超原子価結合について考察する。
1.6kcal/mol、理論値は17.4kcal/molとなり、この分子が熱力学的に安定であることを示している。NaCN分子のイオン化エネルギーは実験値4.920.2ならびに理論値4.66eVを得た。理論計算により、NaCN分子は平面2個直線2個の計4個の安定構造を持ち、最も安定な構造は平面型であることがわかった。前回報告したLiCN分子との比較を混じえ、超原子価結合について考察する。
